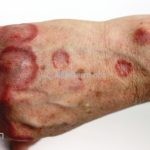
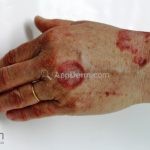
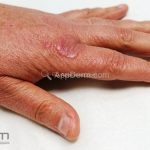
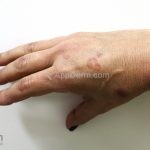
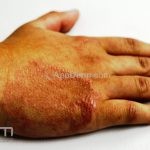
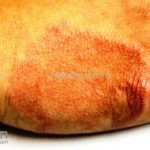
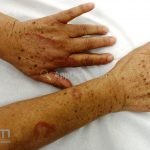
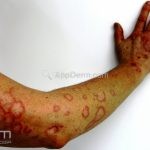
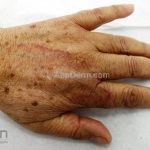
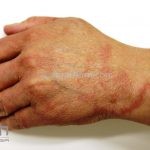
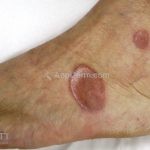
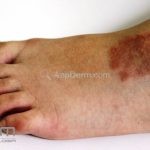
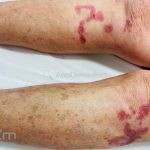
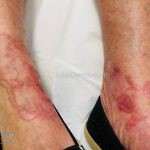
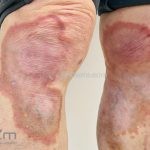
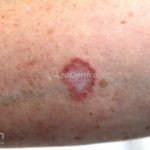
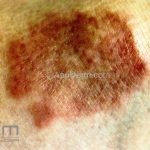
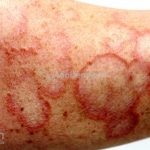
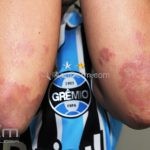
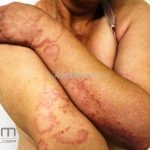
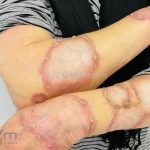
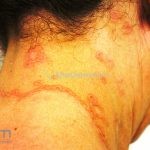
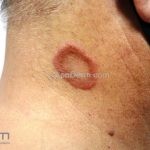
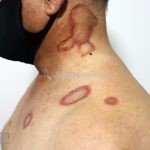
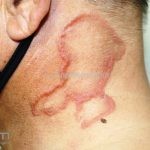
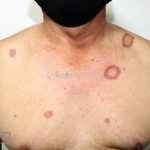
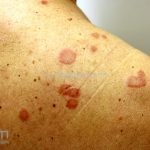
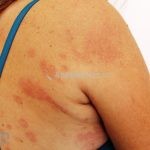
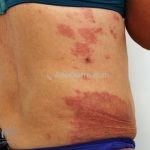
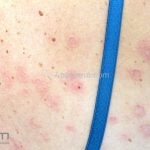
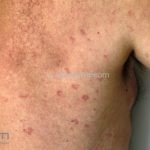
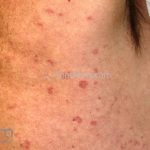
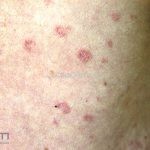
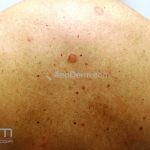
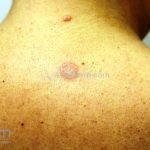
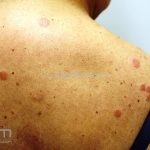
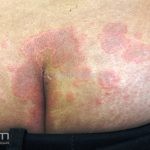
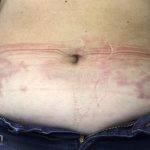
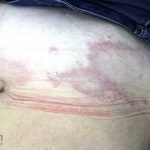
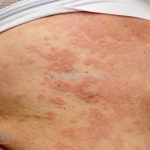
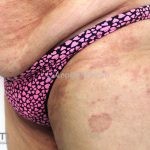
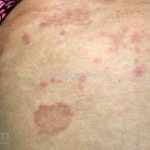
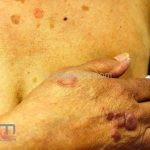
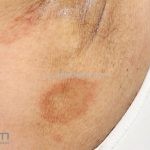
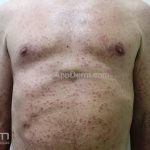
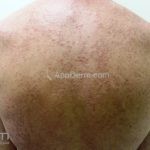
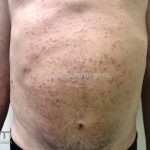
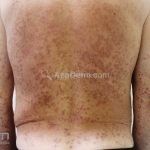
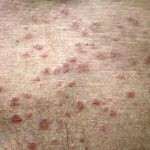
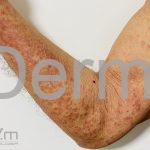
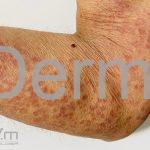
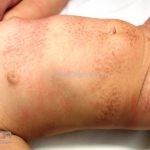
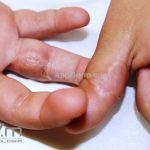
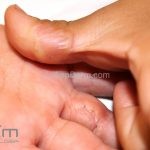
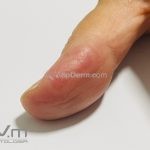
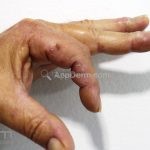
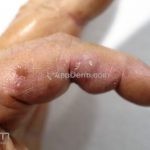
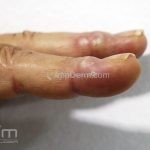
Classicamente, o granuloma anular apresenta-se como placas arciformes a anulares, localizadas nas extremidades de pessoas jovens. As placas podem ter coloração rosa, violácea ou cor da pele. As lesões costumam ser assintomáticas.
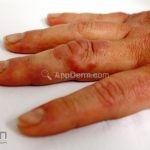
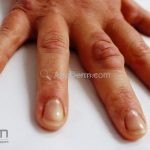
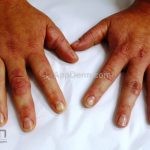
Forma localizada: as lesões são papulonodulares cor da pele ou levemente eritematosas, com 1-3mm de diâmetro, ao coalescer, as lesões assumem o formato anular característico da doença. As lesões crescem de maneira centrífuga e podem apresentar discromias residuais. Os locais mais acometidos são o dorso das mãos, dos dedos, e dos pés, além das superfícies extensoras dos membros.
Forma generalizada (disseminada): ocorre em uma minoria de pacientes. É caracterizada por pápulas normocrômicas ou eritematosas de distribuição simétrica no tronco e extremidades. Essa forma tem inicio tardio, é associada ao HLA-Bw35 e pode estar associada com doenças internas.
Forma subcutânea (profunda): são nódulos grandes, indolores e da cor da pele. Costuma ocorrer em crianças com menos de 6 anos de idade. As mãos, os pés, nádegas, face anterior na tíbia, o couro cabeludo e as pálpebras são os locais de acometimento. 50% dos pacientes apresentam a forma clássica associada.
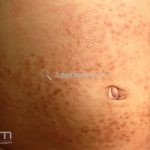
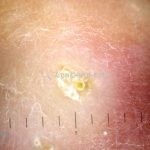
Forma perfurante: múltiplas pápulas eritematosas e descamativas com umbilicação central. Ocorrem no dorso das mãos e dedos. Ao histopatológico, há eliminação transepidérmica de colágeno degenerado. Essa forma pode ocorrer sobre cicatrizes de herpes-zóster.
Forma actínica: descrita por alguns autores. As lesões ocorrem somente em áreas de exposição solar, tem sido associadas à infecção pelo HIV.
Forma eritematosa: descrita em 1979. Observado em pessoas idosas.
Obs: as formas clássica e perfurante podem ocorrer sobre cicatrizes de herpes-zóster.
AppDerm®