Epidermólise bolhosa adquirida
Etiologia:
Doença bolhosa adquirida associada à autoimunidade contra o colágeno tipo 7 (fibrilas de ancoragem).
Associação com HLA-DRB1*1501 e DR5 (em brancos e afro-americanos) e DRB1*13 (em coreanos).
Doenças associadas: doença de Crohn, lúpus eritematoso sistêmico, artrite reumatoide, tireoidites, diabete, etc.
AppDerm®
Dados Epidemiológicos:
Doença rara.
É mais comum em pacientes adultos.
AppDerm®
Manifestações Clinicas:
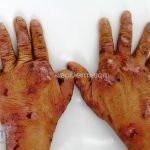
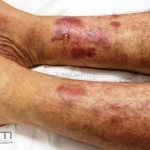
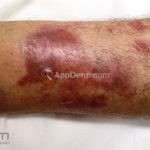
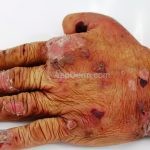
A doença costuma se manifestar na forma mecanobolhosa não inflamatória, caracterizada pelo surgimento de bolhas acrais que se curam com cicatrização atrófica, mília e hiper ou hipopigmentação.
As bolhas e as erosões, que surgem em pele não inflamada ou em áreas de cicatrização, devem ser aquosas, ou com menos frequência, hemorrágicas.
Os locais propensos a traumas são os mais acometidos, especialmente, cotovelos, joelhos e dorso das mãos.
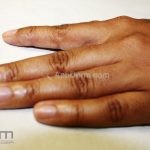
O acometimento acral pode ser mutilante, levando a deformidades dos dedos, sindactilia, distrofia e perda completa das unhas.
O couro cabeludo é acometido em 20% dos pacientes e pode resultar em alopecia cicatricial.
As mucosas podem ser acometidas. Erosões e vesículas intactas podem ser observadas na cavidade oral, laringe e esôfago, podendo levar à disfagia e estenose laríngea.
O acometimento ocular pode provocar cegueira.
Nas crianças, a doença pode apresentar características da dermatose por IgA linear e penfigoide bolhosos.
A doença apresenta períodos de remissão e exacerbação.
AppDerm®
Diagnóstico:
Clínico, histopatológico, imunofluorescência direta (IFD) e Salt Split Skin (SSS).
Histopatológico: bolha com clivagem subepidérmica sem acantólise. A cavidade da bolha costuma estar vazia ou com eritrócitos. O infiltrado inflamatório está ausente ou é mínimo, podendo lembrar o infiltrado do penfigoide bolhoso ou da dermatite herpetiforme.
IFD: depósitos de C3 e IgG ao longo da zona da membrana basal, em um padrão linear.
SSS: a fluorescência ocorre na base (lado dérmico) da bolha.
AppDerm®
Diagnósticos diferenciais:
Penfigoide bolhoso, penfigoide de Brunsting-Perry, porfiria cutânea tarda, pseudoporfirias, epidermólise bolhosa hereditária, lúpus eritematoso sistêmico, amiloidose bolhosa, dermatite herpetiforme e dermatose por IgA linear.
AppDerm®
Tratamento:
Tratamento difícil e normalmente insatisfatório.
Não há protocolo de tratamento.
Imunossupressores (corticoide sistêmico, azatioprina, metotrexato, micofenolato de mofetil, ciclofosfamida).
Dapsona (pode ajudar).
AppDerm®