Doença de Behçet
Etiologia:
É uma doença multissitêmica e polissintomática, que apresenta períodos de atividade e de remissões. Os mecanismos patológicos da doença envolvem danos vasculares e respostas autoimunes.
Neutrófilos e imunocomplexos circulantes parecem ser os responsáveis pelas lesões mucocutâneas.
Na histopatologia, encontramos em reação vascular neutrofílica ou até mesmo uma vasculite leucocitoclástica.
Na doença de Behçet, os neutrófilos produzem uma quantidade aumentada de superóxidos e excesso de enzimas lisossomais, aumentando a quimiotaxia e o dano tecidual.
As evidências epidemiológicas sugerem que fatores genéticos e ambientais contribuem para o desenvolvimento da doença. O alelo HLA-B51 parece ser um importante fator de risco para o desenvolvimento da doença.
Agentes infecciosos associados: Herpes-simples, vírus da hepatite C, parvovírus B19 e Stretptococcus.
AppDerm®
Dados Epidemiológicos:
A doença é mais comum no Japão, no Oriente Médio e na bacia do Mediterrâneo.
A Turquia possui a maior prevalência, 80 : 100.000. Nos EUA, a prevalência é de 0,64 : 100.000. O pico de incidência ocorre entre os 20 e 35 anos de idade.
A forma familiar corresponde de 2 a 5% dos casos, exceto no Oriente Médio, onde representa de 10 a 15%.
AppDerm®
Manifestações Clinicas:
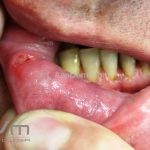
A estomatite aftosa está quase sempre presente durante o curso da doença.
As lesões orais podem preceder as manifestações sistêmicas por muitos anos, mas uma piora dramática na gravidade ocorre em conjunto com o início dos outros achados.
As lesões podem ser indistinguíveis das que ocorrem na aftose complexa.
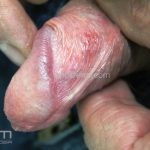
A úlcera genital ocorre no escroto e no pênis (no homem) e na vulva (na mulher).
As úlceras anogenitais tendem a ser maiores que as lesões orais, apresentam margens irregulares e são mais dolorosas. Entre as lesões primárias da doença, estão as vesicopústulas estéreis, acrais e faciais, assim como as pápulas pustulares ou purpúricas.
Mesmo que não sejam foliculocêntricas, as lesões papulopustulosas tem sido descritas como acneiformes.
Lesões que se assemelham com o eritema nodoso costumam acometer as pernas, nádegas, face e o pescoço de mulheres. A patergia pode ocorrer.
Manifestações oculares (principal causa de morbidade): ocorre em 90% dos casos; preferência pele sexo masculino; vasculite retiana (pode causar cegueira); uveíte posterior (achado mais característico) e anterior; glaucoma; conjuntivite; ceratite; esclerite; hemorragia vítrea; e neurite óptica.
Manifestações articulares: artrites mono ou poliarticular não erosivas em 50% dos casos, ocorrem em joelhos, punhos e tornozelos.
Manifestações gastrointestinais: dor; hemorragias; e úlceras gástricas e intestinais (região ileocecal.
Manifestações neurológicas (associadas a um pior prognóstico): meningoencefalite aguda; paralisias de nervos cranianos; lesões no tronco encefálico; e sinais piramidais e extrapiramidais.
Manifestações vasculares: doença oclusiva ou aneurismática; trombose venosa profunda e superficial.
Manifestações cardiopulmonares: arterite coronariana; doença valvar; miocardite; arritimias; e aneurismas de artéria pulmonar.
Manifestações renais: glomerulonefrite. Síndrome MAGIC (mouth and genital ulcers with inflamed cartilage): policondrite recidivante e doença de Behçet.
AppDerm®
Diagnóstico:
Clínico (através dos critérios do International Study Group para o diagnóstico da doença de Behçet):
Critério principal (obrigatório): ulcerações orais recorrentes (três ou mais episódios por ano).
Critérios secundários (dois): ulcerações genitais recorrentes (úlcera genital aftosa ou cicatriz); lesões oculares (uveíte anterior ou posterior, vasculite retiniana, células no vítreo); lesões cutâneas (lesões tipo eritema nodoso e pseudofoliculite); teste de patergia (interpretados em 24-48 horas).
Histopatológico: pode ser inespecífico. A vasculopatia cutânea pode acometer qualquer calibre dos vasos na derme e hipoderme.
Pode se apresentar como uma reação vascular neutrofílica caracterizada pelo infiltrado neutrofílico angiocêntrico, com leucocitoclasia e extravasamento de hemácias, ou como uma vasculite leucocitoclástica com ou sem trombose e necrose de parede.
Nas lesões “tipo eritema nodoso”, os achados podem variar de uma paniculite lobular neutrofílica a uma paniculite lobular e septal com infiltrado inflamatório misto.
AppDerm®
Diagnósticos diferenciais:
Pênfigo vulgar, herpes simples, eritema nodoso, síndrome de Sweet
AppDerm®
Tratamento:
DOENÇA MUCOCUTÂNEA LEVE A MODERADA: Cochicina 0,6 mg, VO, 3x/dia (nível 1 de evidência); Dapsona 50-150 mg, VO, 1x/dia (nível 1 de evidência); Corticoides tópicos e inalatórios (nível 2 de evidência); Corticoides intralesionais (nível 3 de evidência); Lidocaína viscosa, sucralfato tópico e outros tratamentos sintomáticos (nível 3 de evidência); Combinação de dapsona e cochicina oral (nível 3 de evidência).
DOENÇA MUCOCUTÂNEA GRAVE: Talidomida 50-150 mg, VO, à noite (nível 1 de evidência); Inibidores TNF-∂ (nível 1 de evidência); Prednisona intermitente com redução gradual, dose inicial de 40-80mg/dia (nível 2 de evidência); Metotrexato 2,5-25 mg/semanalmente, VO ou IM (nível 3 de evidência).
DOENÇA SISTÊMICA: Prednisona 60-120 mg/dia, VO (nível 2 de evidência); Acetato de metilprednisolona 40 mg, IM, 3x/semana (nível 2 de evidência); Azatioprina 50-100 mg, VO, 2x/dia (nível 1 de evidência); Clorambucil 4-6 mg, VO, 1x/dia (nível 1 de evidência); Tacrolimo 0,1-0,2 mg/kg/dia, VO (nível 2 de evidência); Inibidores do TNF-∂ (nível 2 de evidência). Micofenolato de mofetil 1-1,5 g, VO, 2x/dia (nível 3 de evidência);
AppDerm®