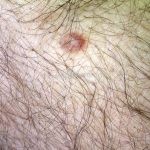
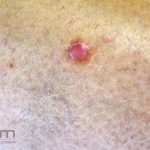
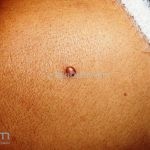
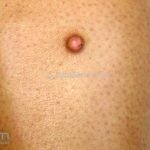
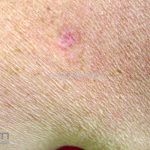
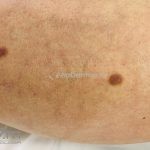
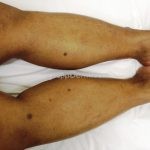
Clínico, dermatoscopia, histopatológico, imuno-histoquímica.
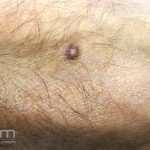
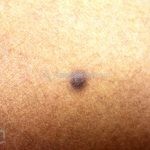
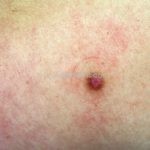
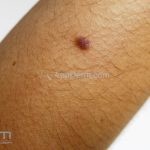
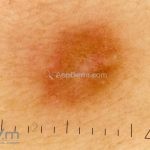
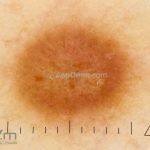
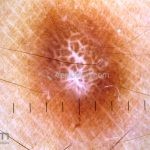
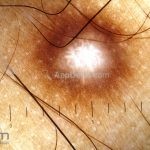
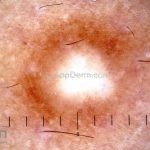
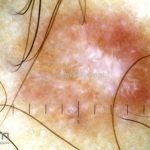
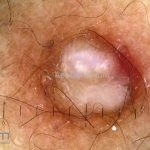
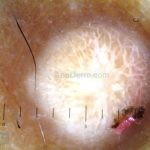
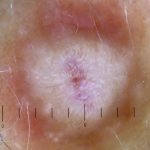
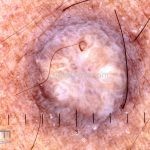
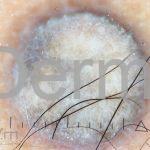
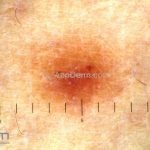
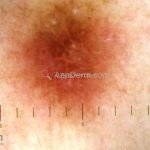
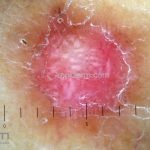
Dermatoscopia (clássica): apresenta rede pigmentar delicada na periferia da lesão e uma placa branca cicatricial central, pode haver pontos e glóbulos castanhos e vasos puntiforme.
Existem nove padrões descritos na dermatoscopia e, muitas vezes, o aspecto é indistinguível do melanoma.
Histopatológico: na epiderme, encontramos acantose com confluência dos cones epiteliais e pigmentação da camada basal (aspecto de “dedos sujos”).
A derme papilar não é envolvida (zona Grenz).
É uma lesão não encapsulada constituída de células fusiformes em padrão fascicular ou estoriforme com colágeno aprisionado, mais notável na periferia da lesão.
Linfócitos, células xantomatosas, células multinucleadas e células pleomórficas podem ser encontradas na mesma lesão.
Variantes histológicas: celular (histiocitoma fibroso); de poucas células (fibroso, hialinizante, queloidiano); com proeminente componente vascular (“hemangioma” esclerosante); aneurismático (hemorrágico ou hemossiderótico); mimetizando hemangiopericitoma; xantomatoso; colesterótico; liquenoide, erosivo e ulcerado; mixoide; atrófico; subcutâneo; combinado; com células atípicas (com células “monstras” ou psedossarcomatoso);
com células gigantes tipo-osteclastos; com calcificação ou ossificação metaplásica; em paliçada; de célula granular; de células claras; de células em anel de sinete; de células balonizantes; com células epitelioides; com folículos linfoides; com infiltrado eosinofílico; com elastofagocitose; com folicular e/ou sebácea comunicação; com diferenciação miofibroblástica; com proliferação de fibras nervosas; adenodermatofibroma; e histiocitoma fibroso metastático. Imuno-histoquímica: positividade para o fator XIIIa, Stromelysin-3, actina músculo-específica e CD68.
AppDerm®