Síndrome de Gardner
Etiologia:
Genodermatose caracterizada por polipose intestinal pré-maligna e adenocarcinoma do trato gastrointestinal.
Ocorre devido a mutação no gene APC, um supressor tumoral.
AppDerm®
Dados Epidemiológicos:
Herança autossômica dominante.
Incidência de 1:8.000 a 1:16.000 nascimentos.
Afeta homens e mulheres.
O risco de malignização dos pólipos intestinais pode chegar a 100%.
AppDerm®
Manifestações Clinicas:
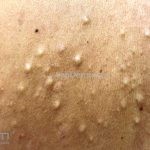
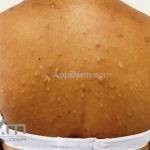
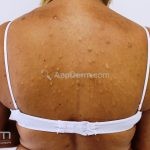
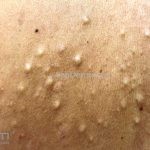
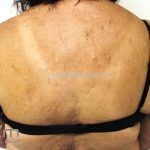
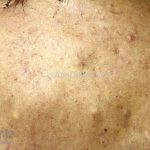
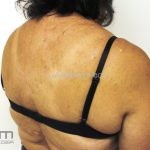
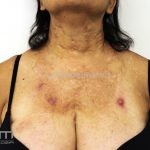
A síndrome é caracteriza-se pela tríade: polipose intestinal pré-maligna , osteomas e múltiplos cistos epidérmicos.
Na pele, os cistos epidérmicos ocorrem precocemente e acometem 50 a 60% dos pacientes, costumam acometer a face, couro cabeludo e extremidades.
Lipomas, leiomiomas, neurofibromas, tricoepiteliomas e cistos ovarianos também foram descritos na síndrome.
Os pólipos adenomaosos pré-malignos são encontrados principalmente no cólon, mas podem ser encontrados em intestino delgado e estômago.
Os osteomas, presentes em 80% dos pacientes, são mais encontrados em mandíbulas, maxilas, crânio e ossos longos.
Exostoses, endostoses e espessamento cortical de ossos longos também podem ocorrer.
Anomalias dentárias ocorrem em 20% dos pacientes, incluem dentes supranumerários, odontomas, dentes inclusos e múltiplas cáries.
Hipertrofia congênita do epitélio pigmentar da retina é visto em cerca de 2/3 dos pacientes e pode ser um sinal precoce da síndrome.
Pacientes com a síndrome possuem um maior risco para o desenvolvimento de outros tumores como o carcinoma papilar de tireóide, hepatoblastoma (em crianças), carcinomas pancreático e de via biliar, adenoma de adrenal, sarcomas, etc.
AppDerm®
Diagnóstico:
Clínico, radiológico, colonoscopia e endoscopia.
AppDerm®
Diagnósticos diferenciais:
Síndrome de Cowden, síndrome de Birt-Hogg-Dubé, síndrome de Muir-Torre, síndrome de Peutz-Jeghers, estatocistomas múltiplos.
AppDerm®
Tratamento:
Colectomia preventiva é recomendada.
As lesões cutâneas podem ser retiradas com pequenas cirurgias.
Realizar colonoscopias anuais e endoscopia digestiva alta a cada 3 anos.
Tratar possível malignização.
Membros da família devem ser avaliados.
AppDerm®