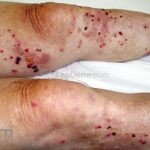
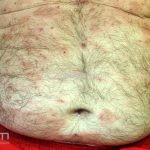
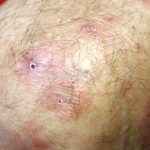
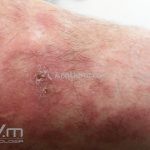
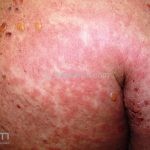
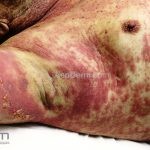
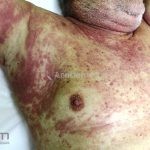
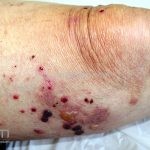
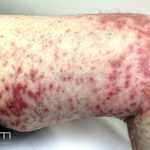
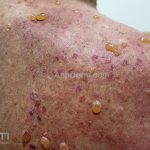
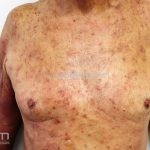
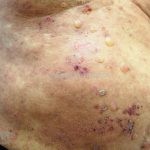
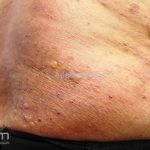
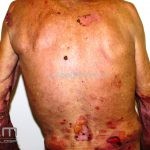
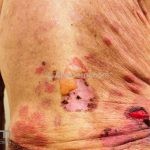
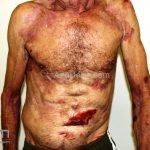
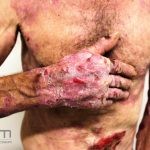
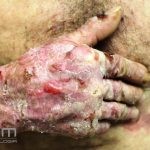
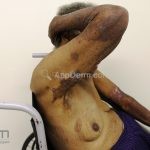
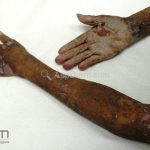
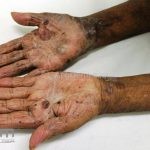
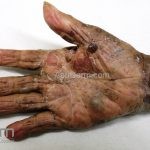
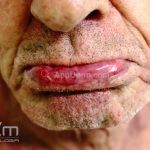
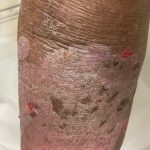
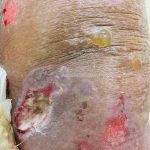
Doença bolhosa autoimune subepidérmica.
Ocorrem respostas humoral e celular contra as moléculas
BPAg2 (180kD) e BPAg1 (230kD) dos hemidesmossomos.
O BPAg1 (230kD) localiza-se na placa hemidesmossômica, enquanto o BPAg2 (180kD) caracteriza-se por uma glicoproteína transmembranosa, cujo domínio extracelular ultrapassa a lâmina lúcida da zona da membrana basal, correspondendo aos filamentos de ancoragem (colágeno XVII).
Doenças associadas: neoplasias malignas (trato digestivo, bexiga, pulmões, linforoliferativas), doença inflamatória intestinal, artrite reumatoide, dermatomiosite, tireoidite de Hashimoto, lúpus eritematoso, trombocitopenia autoimune, psoríase, líquen plano, doença de Parkinson, demência, transtornos psiquiátricos e acidentes vasculares encefálicos.
Outros fatores associados: medicamentos (furosemida, espironolactona, D-penicilamina, captopril, amoxicilina, ciprofloxacina, iodeto de potássio, ouro, analgésicos, etc.), trauma, radioterapia, queimadura, irradiação UV (incluindo PUVA).
Em caucasianos, há uma associação significativa com o alelo DQB1*0301, enquanto na população japonesa a associação ocorre com os alelos DRB1*04, DRB1*1101 e DQB1*0302.
AppDerm®