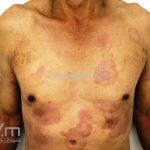
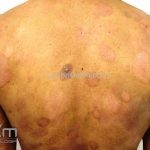
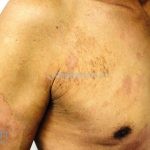
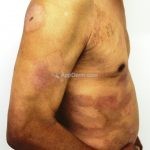
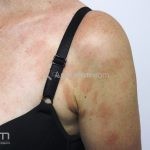
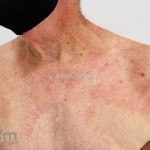
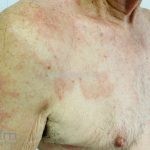
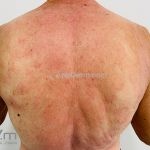
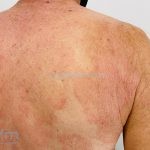
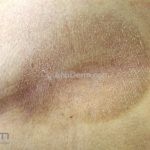
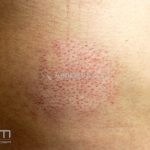
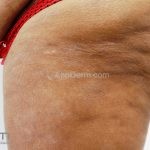
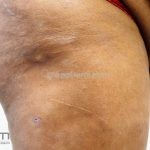
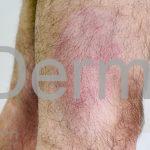
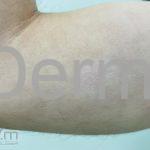
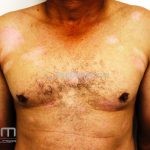
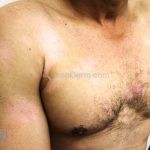
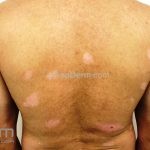
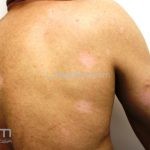
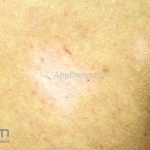
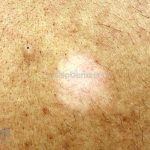
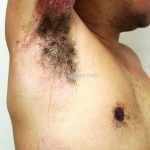
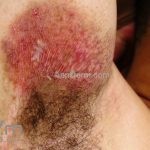
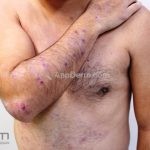
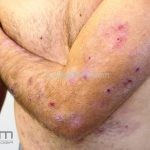
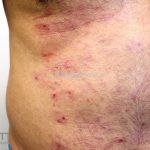
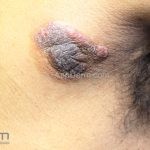
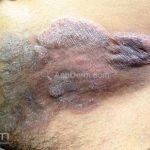
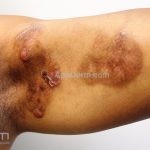
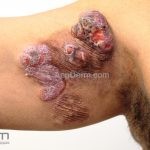
Forma clássica (CD2+, CD3+, CD4+, CD5+, CD8-, CD45RO+, TCR+, CD30-, com frequente perda dos antígenos CD7 e CD26): caracterizada por lesões tipo “patches” (máculas eritematosas, hipercrômicas ou hipocrômicas, dascamativas ou não); e lesões tipo placas (eritematosas, descamativas e infiltradas); nas formas mais agressivas, as lesões tumorais poderão ser encontradas em face e zonas flexurais.
A doença costuma evoluir lentamente, apenas uma pequena porcentagem de pacientes evolui com lesões tumorais, e uma porcentagem ainda menor morre em decorrência dos efeitos sistêmicos do linfoma. A micose fungoide pode iniciar-se em qualquer estágio da doença, e variantes atípicas podem coexistir com as formas típicas da doença.
Poucos pacientes evoluirão para a fase leucêmica da doença, a síndrome de
Sézary ou para linfoma de grandes células.
Forma foliculotrópica (CD3+, CD4+, CD8-, sendo comum a presença de células blásticas CD30+): é caracterizada pela presença do infiltrado linfocítico atípico com tropismo pelo folículo piloso. É considerada uma forma incomum e agressiva de micose fungoide. É a variante associada a mucinose folicular. A sobrevida em 5 anos varia de 70 a 80%.
Forma reticulose pagetoide (CD3+, CD4+, CD8 ou CD3+, CD4-, CD8+, é frequente a expressão de CD30): variante de baixo grau de malignidade, pode ser unilesional (descrita por Woringer-Kolopp) ou disseminada (descrita por Ketron-Goodman). Atualmente, o termo reticuloide pagetoide é utilizado apenas para a forma localizada. Os casos disseminados são classificados como linfoma primário cutâneo agressivo epidermotrópico de células T citotóxica CD8+, LCCT gama/delta+ ou micose fungoide tumoral.
Clinicamente, a reticuloide pagetoide se manifesta como uma placa solitária hiperqueratótica, eritemato-descamativa ou verrucosa. Em geral, localizada em extremidades inferiores de adultos jovens.
A evolução é lenta e progressiva. Disseminação extracutânea ou óbito nunca foram relatados.
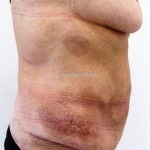
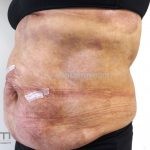
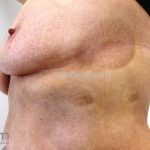
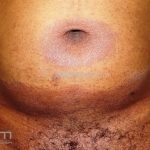
Forma cútis laxa granulomatosa (CD4+, CD8 e CD45RO+, também são relatados CD30, CD3, CD5 e CD7): é rara, caracterizada pelo desenvolvimento lento de pele laxa em dobras flexurais. O envolvimento extracutâneo é pouco frequente, sendo a linfoadenite granulomatosa a manifestação mais comum.
A doença não oferece risco de vida, porém pode progredir e ser acompanhada de uma doença linfoproliferativa.
AppDerm®