Esclerose sistêmica
Etiologia:
Doença autoimune do tecido conjuntivo de etiologia desconhecida.
As principais anormalidades patogênicas da pele e dos órgãos internos são disfunção vascular, ativação imune com produção de anticorpos e fibrose tecidual, caracterizada pela deposição de colágeno e outras proteínas de matriz extracelular.
Disfunção vascular: a angiogênese prejudicada é um dos eventos iniciais da doença, podendo acometer desde pequenos capilares em prega ungueal até grandes vasos pulmonares.
Alterações de células musculares lisas circulantes modificam a produção de fatores vasoconstritor (ex. frio, endotelina) e vasodilatador (ex. óxido nítrico).
O fenômeno de Raynaud e úlceras digitais ocorrem devido a vasoespasmos reversíveis.
Crises renais e pulmonares são manifestações de disfunções de grandes vasos.
Anormalidades estruturais ocorrem com a proliferação da íntima, oclusão luminal, hipóxia, ativação dos fibroblastos e produção do colágeno.
Disfunção imunológica: os pacientes apresentam, predominantemente, citocinas no padrão TH2 (IL-4 e IL-13), que resulta na produção de imunoglobulinas.
Os anticorpos são demonstrados em até 90% dos pacientes com a doença e podem ser usados como indicadores de prognóstico.
Cinco tipos de anticorpos são identificados e relacionados diretamente com a esclerose sistêmica: anti-SCL70 (antitopoisomerase I), anticentrômero, antinucleolar, anti-Fc gama e antimitocondrial.
Anticorpos anti-SCL-70 (topoisomerase I ou antiescleroderma 70): observado na forma difusa da doença, indicando um prognóstico ruim. Apresenta um padrão pontilhado para anticorpos antinucleares, presente em 20-30% dos pacientes.
Anticorpos anticentrômero: observado na forma limitada da doença (sd. CREST), indica um prognóstico bom.
Anticorpos antinucleolar: ocorrem em 50% dos pacientes com doença sistêmica. Os principais são U3-RNP, Rna polimerase, Th/To e PM-Scl. O U3-RNP (fibrilarina) está relacionados com doença sistêmica severa, acometimento cutâneo difuso e intensas telangiectasias. O PM-Scl está associado a polimiosite com esclerodermia. Anti-Th/To é considerado um marcador inicial de doença sistêmica.
Anticorpos anti-receptores FC gama: pode ocorrer no LES, na síndrome de Sjögren, além de outras doenças autoimunes.
Anticorpos antimitocondrial: associação com cirrose biliar primária, tireoidite de Hashimoto, lúpus eritematoso e artrite reumatoide. Não possui valor diagnóstico, mas quando presente pode indicar doença sistêmica ou associação com outras doenças autoimunes.
Fibrose: representa a via final comum na esclerose sistêmica, ocorre deposição excessiva de colágeno, proteoglicanos, fibronectina, fibrilinas e moléculas de adesão (ß1-integrinas).
Desencadentes ocupacionais: derivados do vinil, solventes orgânicos, pesticidas, epóxi e sílica.
Desencadeantes iatrogênicos: bleomicina, cocaína, silicone, óleos tóxicos e partazocine.
AppDerm®
Dados Epidemiológicos:
Distribuição universal, acomete todas as raças.
As mulheres são mais afetadas (3-4 : 1).
A doença costuma ocorrer entre 30 e 50 anos de idade, mas pode acometer crianças e idosos.
Pacientes negros podem apresentar início precoce da doença e um pior prognóstico.
1,5% dos pacientes apresentam história familiar positiva, aumentando, em 10 a 15%, o risco de desenvolvimento da doença.
AppDerm®
Manifestações Clinicas:
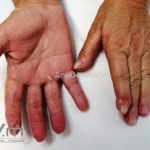
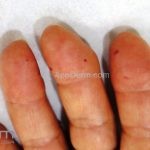
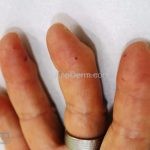
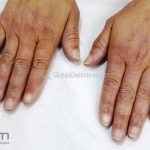
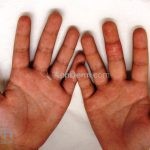
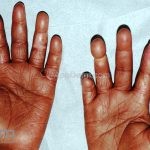
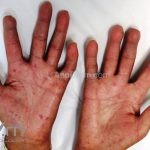
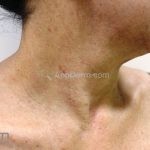
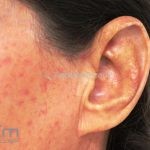
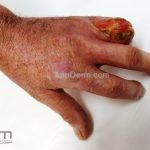
O fenômeno de Raynaud e o surgimento de telangiectasias nas pregas ungueais costumam ser as manifestações iniciais da esclerose sistêmica.
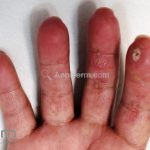
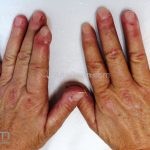
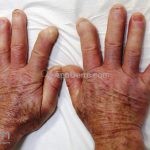
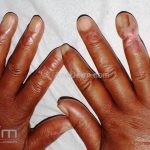
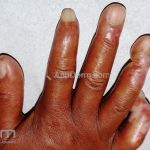
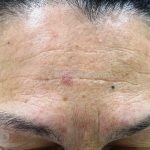
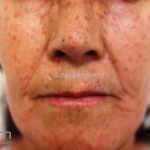
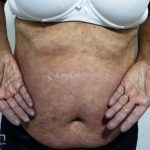
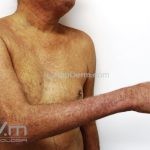
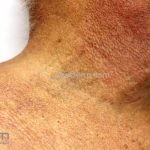
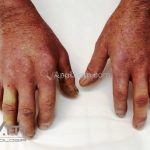
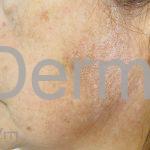
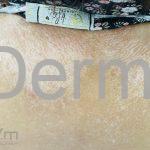
As lesões cutâneas iniciais de esclerose sistêmica podem ser indistinguíveis das lesões da morfeia, tanto no aspecto clínico como no histopatológico. A pele posteriormente endurece e desenvolve uma aparência brilhante e tensa. Os dedos podem desenvolver contraturas em flexão e úlceras, enquanto o envolvimento do rosto pode levar a um nariz de adunco, microstomia e uma aparência jovem.
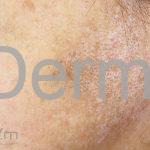
Na pele, além da fibrose, comumente, observamos despigmentação em áreas de fibrose. Alguns pacientes manifestam hiperpigmentação difusa, com acentuação em áreas expostas ao sol e locais de pressão. A leucodermia é caracterizada por áreas localizadas de despigmentação, poupando a pele perifolicular, também conhecida como o sinal de “sal e pimenta”, uma ferramenta útil para o diagnóstico. O pigmento também pode se acumular no trajeto de veias superficiais. Essa leucodermia é mais comum na parte superior do tronco e parte central do rosto.
A pele fica seca em virtude da diminuição da sudorese e o prurido pode ser acentuado.
A calcinose cutânea distrófica costuma acometer as articulações distais das extremidades.
Com base no envolvimento da pele, existem dois subtipos principais: limitado e difuso.
Esclerose sistêmica cutânea limitada: doença cutânea limitada às extremidades distais e ao rosto. O prognóstico é bom.
Os pacientes podem apresentar: fenômeno de Raynaud (99%), inchaço no dedo (90%), artralgia (90%), fricção no tendão (5%) fraqueza proximal (60%), calcinose (40%), telangiectasias (90%), capilares em prega ungueal (>90%), dismotilidade esofágica (90%), envolvimento do intestino delgado (60%), doença pulmonar intersticial (35%), hipertensão pulmonar (20-25%), cardiomiopatia (10%), crise renal (1%), síndrome de Sjögren/Sicca (35%), FAN positivo (90%), anticentrômero positivo (50-90%), anti–Scl-70 positivo (10-15%), sobrevida em 5 anos (90%), sobrevida em 10 anos (70%).
O acrônimo síndrome CREST descreve as características clínicas de um subgrupo de pacientes que apresentam Calcinose, fenômeno de Raynaud, Esofagopatia, eSclerodactilia e Telangiectasias.
Esclerose sistêmica cutânea difusa: acomete extremidades distais e proximais, o rosto e o tronco. O prognóstico é ruim.
Os pacientes podem apresentar: fenômeno de Raynaud (90%), inchaço no dedo (95%), artralgia (98%), fricção no tendão (70%) fraqueza proximal (80%), calcinose (20%), telangiectasias (60%), capilares em prega ungueal (>90%), dismotilidade esofágica (80%), envolvimento do intestino delgado (40%), doença pulmonar intersticial (70%), hipertensão pulmonar (15-20%), cardiomiopatia (15%), crise renal (20%), síndrome de Sjögren/Sicca (15%), FAN positivo (90%), anticentrômero positivo (5-30%), anti–Scl-70 positivo (20-60%), sobrevida em 5 anos (70%), sobrevida em 10 anos (50%).
AppDerm®
Diagnóstico:
A orientação diagnóstica para a doença estabelecida é baseada nos critérios do Colégio Americano de Reumatologia (American College of Rheumatology – ACR), que classifica o paciente com a doença na presença do critério maior ou pelo menos dois dos critérios menores:
Critério maior: fibrose simétrica da pele proximal às metacarpofalangianas ou
metatarsofalangianas.
Critérios menores (2 ou mais): esclerodactilia, úlceras ou microcicatrizes ou perda de substância das polpas digitais, fibrose pulmonar bilateral.
Embora apresentem altas sensibilidade e especificidade diagnósticas, tais critérios não detectam, adequadamente, pacientes com doença inicial.
Neste sentido, LeRoy e Medsger propuseram os seguintes critérios para o diagnóstico de formas iniciais de esclerose sistêmica: evidência objetiva (observada pelo médico) de fenômeno de Raynaud associado a capilaroscopia periungueal ou autoanticorpos específicos para a doença (anticentrômero, antitopoisomerase I, antifibrilarina, anti-PM-Scl); ou evidência subjetiva (à anamnese) de fenômeno de Raynaud mais padrão esclerose sistêmica à capilaroscopia periungueal e
autoanticorpos específicos para esclerose sistêmica.
Histopatológico: encontramos espessamento do colágeno, com diminuição das fendas entremeadas ás fibras, preenchendo a derme e estendendo-se aos septos fibrosos do tecido adiposo; perda da gordura periécrina; perda de estruturas anexiais (casos avançados); além de edema e inflamação perivascular linfoplasmocitária (em lesões iniciais).
AppDerm®
Diagnósticos diferenciais:
Morfeia, dermatomiosite, doença mista do tecido conjuntivo, lúpus eritematoso sistêmico, escleroedema, escleromixedema, fasciíte eosinofílica, fibrose nefrogênica sistêmica, doença enxerto versus hospedeiro.
AppDerm®
Tratamento:
O tratamento é difícil e multidisciplinar.
A abordagem terapêutica tem como objetivo controlar os sintomas e aumentar a sobrevida dos pacientes. Abordaremos os tratamentos de acordo com os achados clínicos.
Fenômeno de Raynaud: manter os membros aquecidos e evitar o tabagismo. Nifedipina (mais utilizada), losartana (também efetiva), sildenafila (resultados mistos), tadalafila (resultados mistos), AAS (empírico) e pentoxifilina (empírico) podem ser usados.
Esclerose cutânea: evolui independente do tratamento. D-penicilamina e metotrexato (podem ajudar); minociclina (não apresentou resultados); PUVA e UVA1 (podem ser úteis).
Telangiectasias: podem ser tratadas com laser de corante pulsado.
Calcificações: não possui tratamento farmacológico efetivo, cirurgias podem ser necessárias.
Doença pulmonar intersticial: imunossupressão com ciclofosfamida ou micofenolato de mofetil (principalmente).
Hipertensão pulmonar: vasodilatadores, antagonistas dos receptores de endotelina (bosentana, sitaxentana, ambrisentana); análagos da prostaciclina (iloprost/inalado, epoprostenol/EV e teprostinila/SC); e inibidores da PDES (sildenafila, tadalafila).
Cardiomiopatia: diuréticos, inibidores da ECA, ß-bloqueadores, bloqueadores dos receptores da angiotensina II, antagonistas da aldosterona.
Doença renal: inibidores da ECA (importância comprovada, diminui o número de crises renais).
Doença gastrointestinal: inibidores de bomba de prótons (para o refluxo) e domperidona ou metoclopramida (para melhorar a motilidade e o inchaço).
AppDerm®